Research
Human health, development, and disease emerge from complex interactions within and between cells. Faithfully measuring salient features of biological systems is difficult as key interactions are nested across dynamically interacting scales — from omic layers to cellular niches and tissues to organ systems. To address these challenges and drive discovery, the MDL operates as an integrated team of molecular and computational scientists developing and applying robust, scalable methods across many domains.
RNA Isoform Sequencing, Methods & Applications
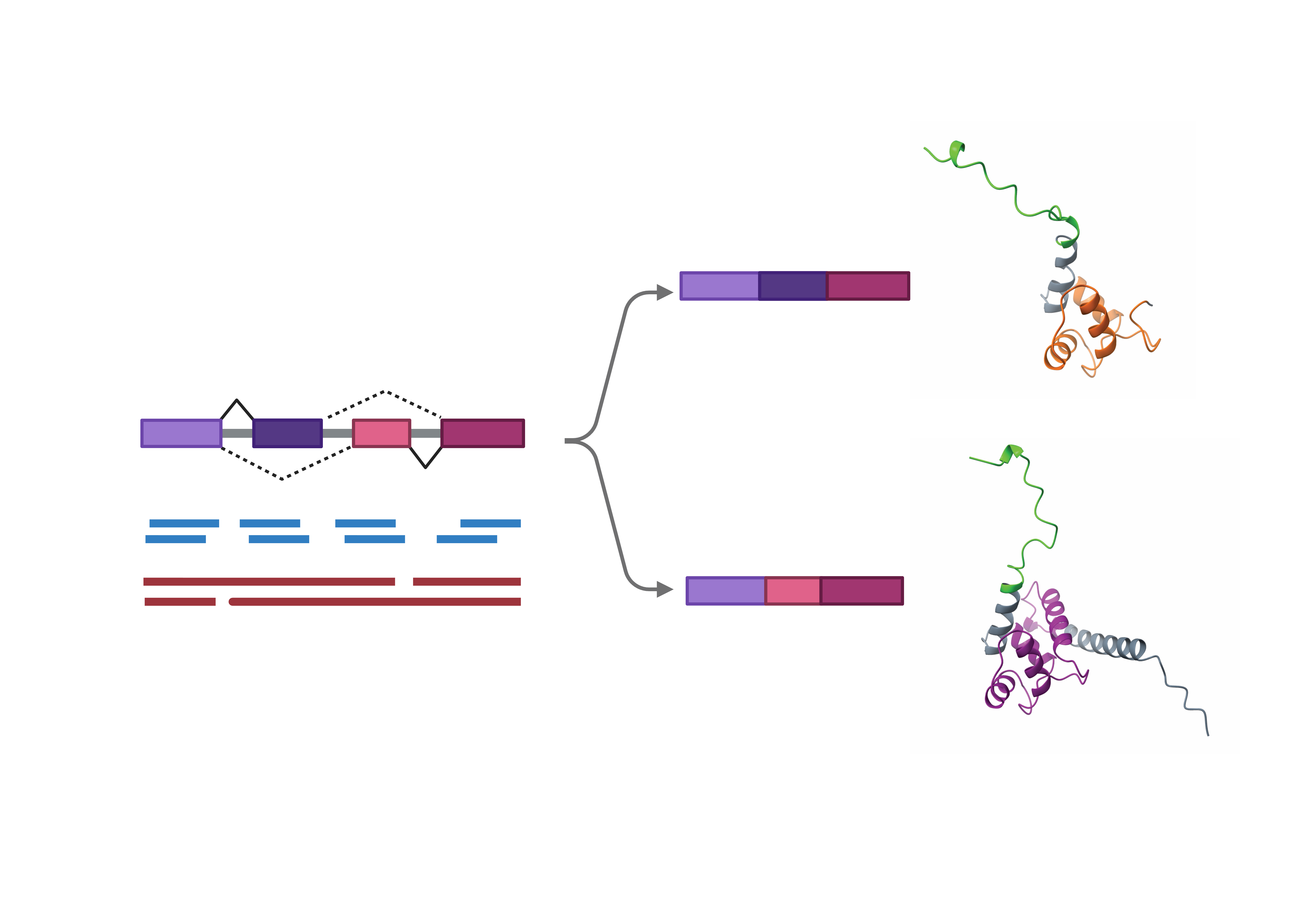
The MDL is actively developing integrated tool sets for long-read RNA isoform sequencing and analysis. Our vision is to bridge systems, molecular, and structural biology through this feature-rich and scalable assay.
Alternative splicing is a core regulatory process that allows a single gene to produce multiple distinct mRNA isoforms, significantly expanding the functional diversity of the human proteome. This mechanism is essential for orchestrating developmental transitions and tissue-specific maturation by fine-tuning proteoform composition. When these splicing programs are disrupted, pathogenic isoforms can drive the progression of diverse diseases, including cancer and neurological disorders. Consequently, identifying the full spectrum of these isoforms provides a powerful foundation for both high-resolution diagnostics and targeted therapeutic development.
Traditional transcriptomics has long relied on short-read sequencing, a method that enables high throughput but is limited by its capture of only fragmented mRNA segments. While effective for quantifying gene expression and individual splice junctions, short reads cannot reliably resolve the full structural connectivity of transcripts, forcing researchers to rely on error-prone computational assembly to infer isoform structure. Consequently, analyses have largely focused on gene-level expression, depending on broad inferences of 'gene function' derived from often distant biological contexts. However, this pragmatic abstraction is fundamentally confounded by the functional diversity of protein isoforms, which can exhibit distinct (even antagonistic) biological roles. Continued reliance on these gene-level abstractions risks compromising advanced modeling efforts. Driven by advancements in long-read sequencing platforms, this compromise is no longer necessary – the next era of transcriptomics will be defined by isoform sequencing and analysis.
MAS-ISO-seq: Increasing long-read RNA sequencing throughput
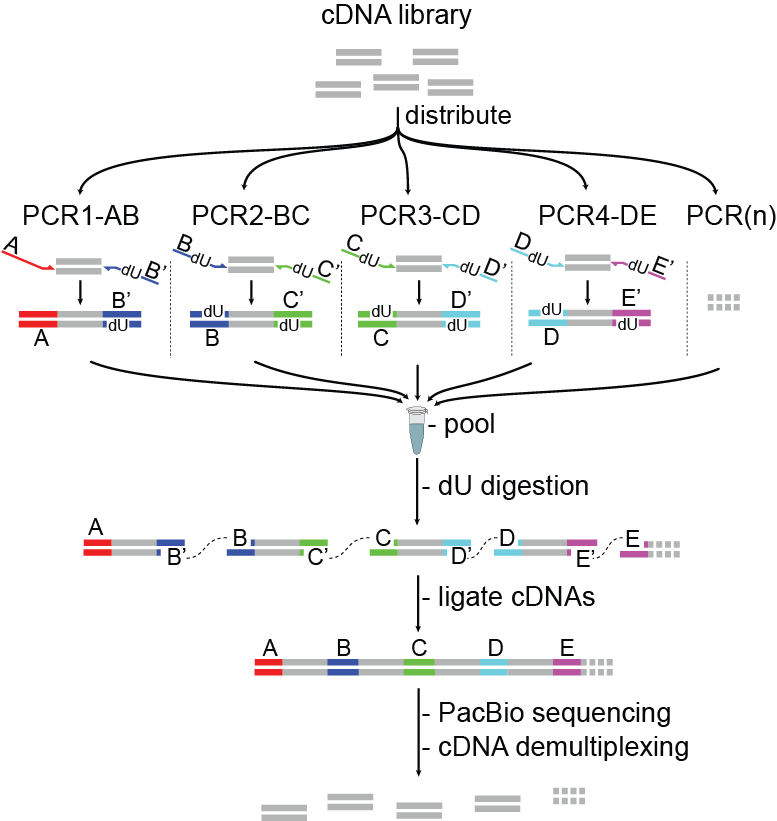
Long-read RNA sequencing enables the detection of complete transcript isoforms, yet its broad application has been constrained by throughput limitations. To address this challenge, we developed multiplexed arrays isoform sequencing (MAS-ISO-seq), a method that programmably concatenates multiple cDNA molecules into single long fragments optimal for long-read sequencers. The workflow creates ordered arrays of cDNAs that are sequenced and subsequently computationally de-concatenated, increasing the sequencing yield 16-fold. This increased throughput enables deep single-cell isoform profiling and significantly improves the identification of novel isoforms and quantification of differentially spliced genes.
CTAT-LR-Fusion: Efficient Transcript Fusion Identification
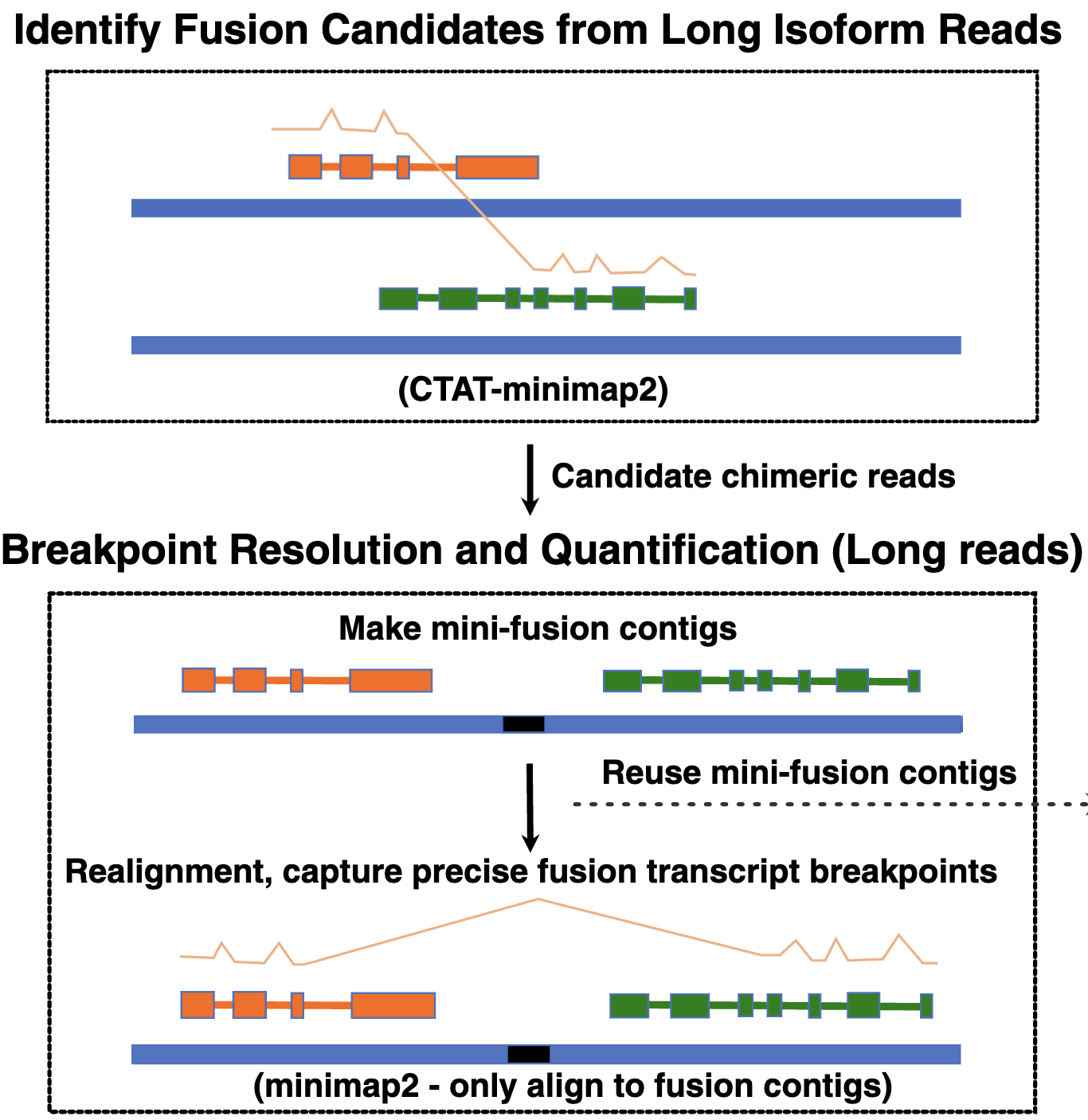
Accurate detection of fusion transcripts is essential for cancer research and diagnostics, but standard short-read sequencing often lacks the length to resolve complete isoforms or detect fusions in 3' or 5’-biased single-cell assays. To overcome these barriers, we developed CTAT-LR-Fusion, a new computational method for identifying fusion transcripts from long-read isoform sequencing in both bulk and single-cell samples. The tool functions by rapidly extracting candidate chimeric reads with a customized aligner, constructing fusion contigs, and integrating optional short-read data to maximize detection sensitivity and breakpoint resolution. Benchmarking on simulated and real cancer datasets confirms that this approach yields higher accuracy than existing methods, revealing complex fusion splicing isoforms and tumor-specific cells missed by short-read analysis. The CTAT-LR-Fusion software is freely available for the research community.
marti: Efficient Artifact Discovery and Quantification

While long-read single-cell transcriptomics enables full-length isoform discovery, accurate analysis is frequently compromised by library preparation artifacts that can be erroneously assigned as novel transcripts. To address this challenge, MDL and Popic Lab co-developed marti, a lightweight software framework designed for the comprehensive classification and quantification of artifactual long-read cDNA constructs. The tool operates by identifying predefined target sequences, such as adapters and primers, within each read to build a structural representation that is then categorized into specific artifact classes or confirmed as a proper read. Application of marti across multiple single-cell platforms revealed distinct artifact profiles—such as high TSO-TSO rates in 10x 3’ libraries—demonstrating its utility in filtering data to ensure high-quality isoform quantification. The marti software and documentation are freely available for the research community at https://github.com/PopicLab/marti.
Billion-scale Single-cell & Perturb-seq
Through illuminating the complex cell types that compose tissues and providing the resolution to pinpoint cellular dysfunction that underlie disease, single-cell RNA-seq has become a cornerstone assay for modern biology. Recent advances in single-cell technology have made million cell scale experiments routine and billion cell flagship projects possible. This dramatic increase in scale now marks a new era in functional genomics, by enabling high-powered assays such as Perturb-seq to systematically dissect genetic determinants of cell function. At MDL, we are leading development efforts at Broad to massively scale up single-cell and Perturb-seq.
More single-cell data will be generated in the next year than the previous ten. This dramatic shift in capability brings front and center the outstanding computational and analytical challenges with this scale of data. The Cellarium team at Broad has been tackling precisely these problems, has now joined MDL. As an integrated team we are building molecular and computational methods to enable the future of single-cell & perturb-seq.
ITV: Integrative Transcriptome Viewer
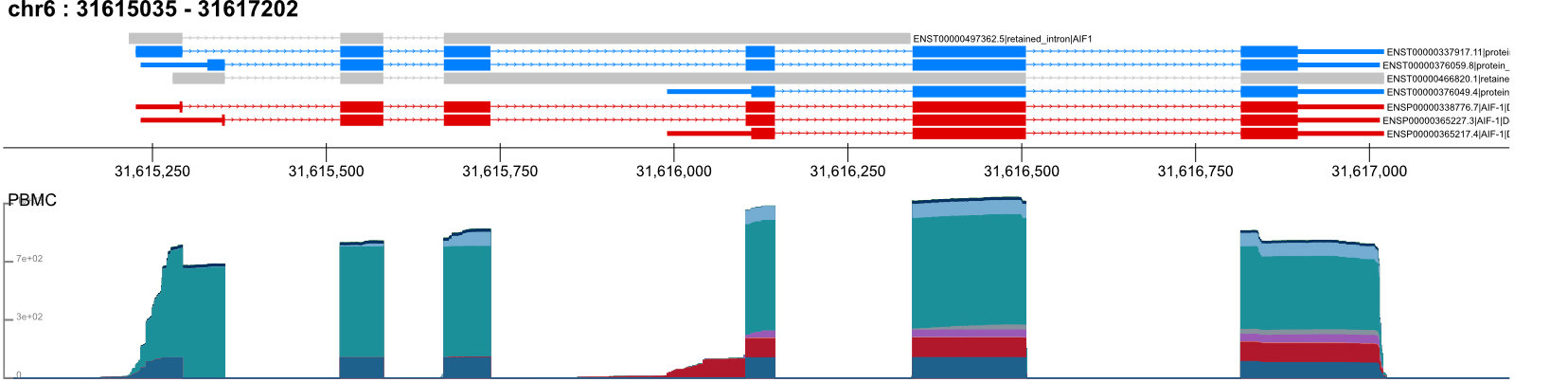
The Integrative Transcriptomics Viewer (ITV) is a Python package developed as a fork of Genomeview to provide specialized visualization capabilities for long-read RNA sequencing data. ITV functions effectively within Jupyter notebook, where it can be run directly in its environment or linked to a preexisting kernel for flexible analysis, but it can also be used through the CLI or scripts for integration with automated pipelines. Software and installation instructions are available for the research community at https://github.com/MethodsDev/ITV and the API is documented on https://itv.readthedocs.io/en/latest/.